Inherited Retinal Disease (IRD) Programs
OPGx-BEST1
OPGx-BEST1 is being developed for bestrophin-1 (BEST1)-related inherited retinal diseases or bestrophinopathies, a form of macular degeneration found primarily in adults.
OPGx-BEST1 is a gene therapy in clinical development for BEST1-related inherited retinal diseases, including Best vitelliform macular dystrophy (BVMD) and autosomal recessive bestrophinopathy (ARB). These forms of inherited macular degeneration primarily affect adolescents and adults and are estimated to impact approximately 8,400 people in the U.S.
Bestrophinopathy is characterized by retinal lesions, with symptoms including dimness of vision, metamorphopsia (distorted vision) or scotoma (blind spot). OPGx-BEST1 leverages an adeno-associated virus (AAV) vector to deliver a functional copy of the BEST1 gene to the retina so that bestrophin-1 protein is produced in retinal pigment epithelial (RPE) cells. This gene therapy approach aims to restore normal function of the RPE cells such that they can provide proper support to photoreceptors, the cells that detect light.
OPGx-BEST1 is administered as a one-time subretinal injection and uses an adeno-associated virus (AAV) vector to deliver functional copies of the BEST1 gene to RPE cells, augmenting the mutated gene and restoring RPE support of photoreceptors. Gene therapies such as OPGx-BEST1 are expected to provide long-lasting benefit, potentially for the lifetime of the patient.
For more information, visit clinicaltrials.gov (NCT07185256).
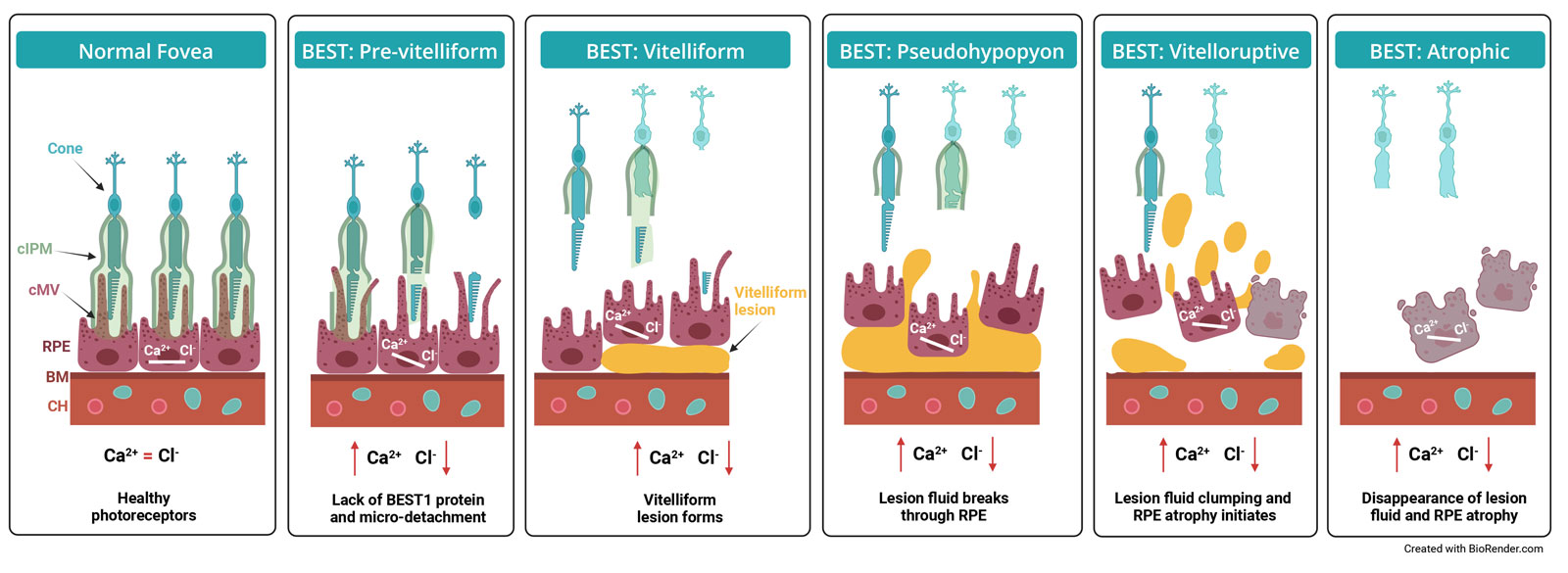 BEST1-related disease is caused by mutations in the BEST1 gene, which encodes bestrophin-1, a calcium-activated chloride channel expressed in retinal pigment epithelial (RPE) cells. Disruption of BEST1 function leads to impaired ionic homeostasis, accumulation of vitelliform (“egg-yolk-like”) material, micro-detachments between the RPE and photoreceptors, progressive RPE atrophy, and eventual central vision loss.
BEST1-related disease is caused by mutations in the BEST1 gene, which encodes bestrophin-1, a calcium-activated chloride channel expressed in retinal pigment epithelial (RPE) cells. Disruption of BEST1 function leads to impaired ionic homeostasis, accumulation of vitelliform (“egg-yolk-like”) material, micro-detachments between the RPE and photoreceptors, progressive RPE atrophy, and eventual central vision loss.
OPGx-LCA5
OPGx-LCA5 is being developed for Leber Congenital Amaurosis Type 5 (LCA5) inherited retinal diseases found in adults and children.
OPGx-LCA5 is a gene therapy in clinical development for Leber congenital amaurosis type 5 (LCA5), a severe, ultra-rare inherited retinal disease affecting approximately 170 people in the U.S. LCA5 is caused by mutations in the LCA5 gene, which encodes lebercilin, a ciliary protein essential for protein trafficking between photoreceptor inner and outer segments. Loss of lebercilin disrupts outer segment development, leading to profound vision loss early in life.
OPGx-LCA5 is designed as a one-time subretinal gene therapy that delivers a functional copy of LCA5 to photoreceptors. Structural–functional dissociation observed in LCA5 patients, where retinal structure may persist despite severe functional loss, suggests a meaningful therapeutic window for intervention.’
OPGx-LCA5 has the potential to become the first approved gene therapy and one-time treatment for LCA5. The program has received a multi-million dollar grant from the FDA Office of Orphan Drug Products as well multiple regulatory designations: Rare Pediatric Disease, Orphan Drug, and Regenerative Medicine Advanced Therapy (RMAT).
For more information, visit clinicaltrials.gov (NCT05616793).
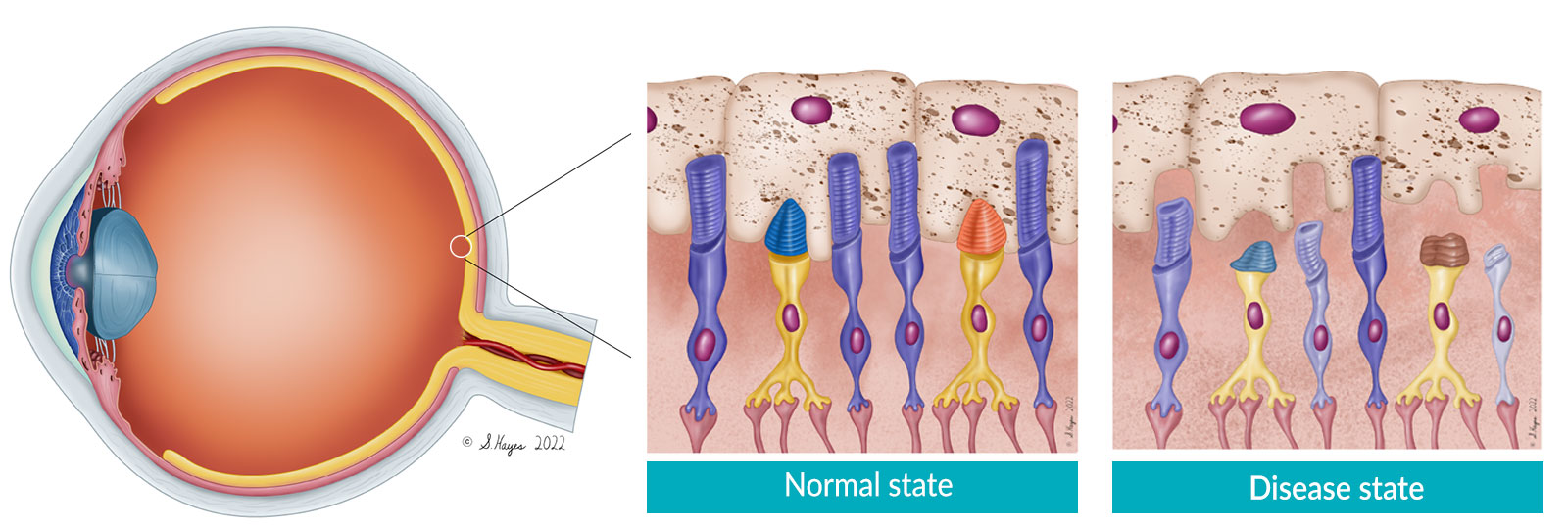
LCA5 is a severe, early-onset form of IRDs. The LCA5 gene encodes for the protein lebercilin, a ciliary protein which is critical for bidirectional protein trafficking in photoreceptor inner and outer segments. Photoreceptors are retinal cells that enable vision by absorbing light and transducing it into an electrochemical signal that is communicated to the visual centers of the brain. In LCA5, the outer segments do not properly develop and photoreceptor function is severely impaired.
OPGx-RHO
OPGx-RHO is a gene therapy that targets autosomal dominant retinitis pigmentosa caused by RHO mutations (RHO-adRP). RHO-adRP is one of the most common IRDs.
OPGx-RHO is a gene therapy program targeting autosomal dominant retinitis pigmentosa caused by mutations in the RHO gene, affecting approximately 8,800 people in the U.S. RHO encodes rhodopsin, a key photopigment in rod photoreceptors essential for light detection and outer segment structure.
OPGx-RHO is designed to preserve rod photoreceptors by replacing the mutant RHO gene with a functional copy, addressing the underlying genetic cause of disease. The program is currently in preclinical development.
OPGx-RDH12
OPGx-RDH12 targets retinal dystrophy caused by mutations in the RDH12 gene, which affects approximately 2,500 people in the U.S. RDH12-associated disease is a severe, early-onset IRD marked by visual acuity loss in early childhood and rapid progression during adolescence.
RDH12 encodes a retinol dehydrogenase enzyme involved in the visual cycle, protecting photoreceptors from toxic metabolite accumulation. OPGx-RDH12 delivers a functional RDH12 gene to photoreceptors using an AAV vector. Preclinical studies in cellular and mouse models have demonstrated restoration of RDH12 activity and functional improvement.
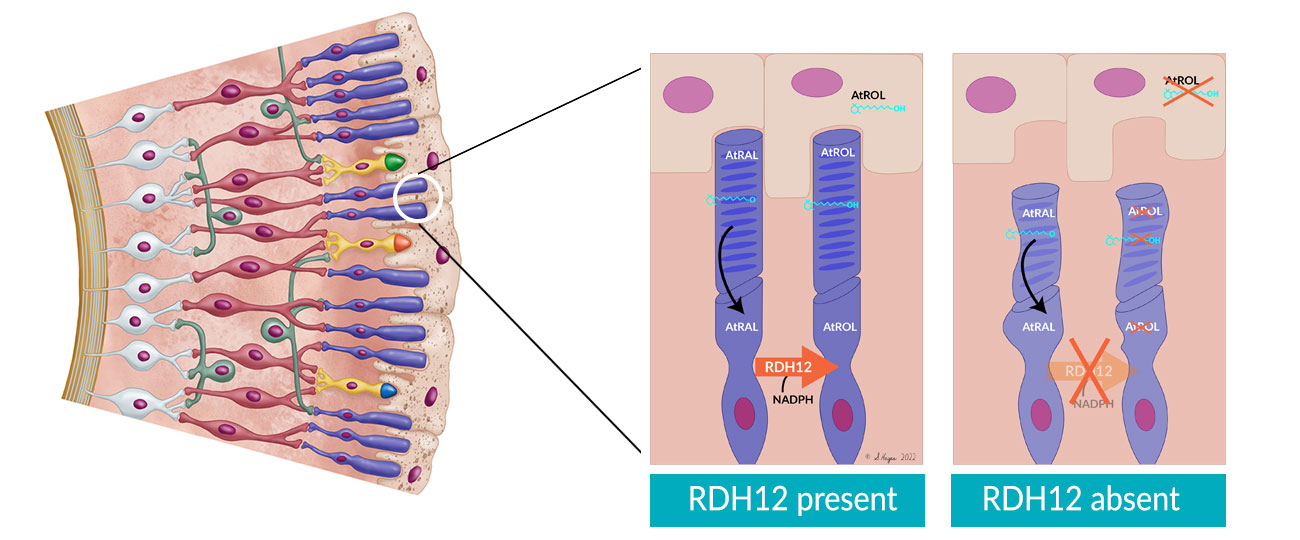
RDH12 is a severe, early-onset IRD. The RDH12 gene encodes for a retinol dehydrogenase enzyme produced in the photoreceptor inner segment that has a key role in the visual cycle. Conversion of all-trans retinal (AtRAL) to all-trans retinol (AtROL) by RDH12 during the visual cycle protects photoreceptors from cytotoxicity. Mutations in RDH12 permit a toxic accumulation of AtROL, leading to photoreceptor degeneration.
OPGx-MERTK
OPGx-MERTK is being developed for retinal degeneration caused by mutations in the MERTK gene, affecting approximately 2,600 people in the U.S. MERTK plays a critical role in phagocytosis of photoreceptor outer segments by RPE cells. Loss of this function leads to rod-cone dystrophy and progressive vision loss.
Opus is advancing OPGx-MERTK using a modern AAV vector design, building on prior preclinical proof of concept and earlier clinical experience, with the aim of improving durability and efficacy.
OPGx-NMNAT1
OPGx-NMNAT1 is a gene augmentation therapy designed to halt disease progression in pediatric patients with retinal degeneration caused by mutations in the NMNAT1 gene, which affects approximately 1,200 people in the U.S.
NMNAT1 is essential for regenerating NAD+, a critical metabolic cofactor. Preclinical data in mouse models that recapitulate key features of the human disease demonstrated that AAV-mediated delivery of NMNAT1 stabilized retinal degeneration.
OPGx-CNGB1
OPGx-CNGB1 is an AAV gene therapy program for a late-onset form of retinitis pigmentosa caused by mutations in the CNGB1 gene, affecting approximately 2,100 people in the U.S.
Opus is collaborating with an NIH-funded academic consortium and the Foundation for the NIH’s Bespoke Gene Therapy Consortium to advance OPGx-CNGB1 into and through a Phase 1 clinical trial.
