Advancing a Diversified Portfolio of First-in-Class Gene Therapies
Inherited Retinal Disease (IRD) Pipeline
OPGx-LCA5
OPGx-LCA5 is being developed for Leber Congenital Amaurosis Type 5 (LCA5) inherited retinal diseases found in adults and children.
Learn more about OPGx-LCA5OPGx-BEST1
OPGx-BEST1 is being developed for bestrophin-1 (BEST1)-related inherited retinal diseases or bestrophinopathies, a form of macular degeneration found primarily in adults.
Learn more about OPGx-BEST1OPGx-RHO
OPGx-RHO is a gene therapy that targets autosomal dominant retinitis pigmentosa caused by RHO mutations (RHO-adRP), which is one of the most common IRDs, affecting approximately 8,800 people in the U.S.
RHO encodes rhodopsin, a key photopigment in rod photoreceptors essential for light detection and outer segment structure. OPGx-RHO is designed to preserve rod photoreceptors by replacing the mutant RHO gene with a functional copy, addressing the underlying genetic cause of disease. The program is currently in preclinical development.
OPGx-RDH12
OPGx-RDH12 targets retinal dystrophy caused by mutations in the RDH12 gene, which affects approximately 2,500 people in the U.S. RDH12-associated disease is a severe, early-onset IRD marked by visual acuity loss in early childhood and rapid progression during adolescence.
RDH12 encodes a retinol dehydrogenase enzyme involved in the visual cycle, protecting photoreceptors from toxic metabolite accumulation. OPGx-RDH12 delivers a functional RDH12 gene to photoreceptors using an AAV vector. Preclinical studies in cellular and mouse models have demonstrated restoration of RDH12 activity and functional improvement.
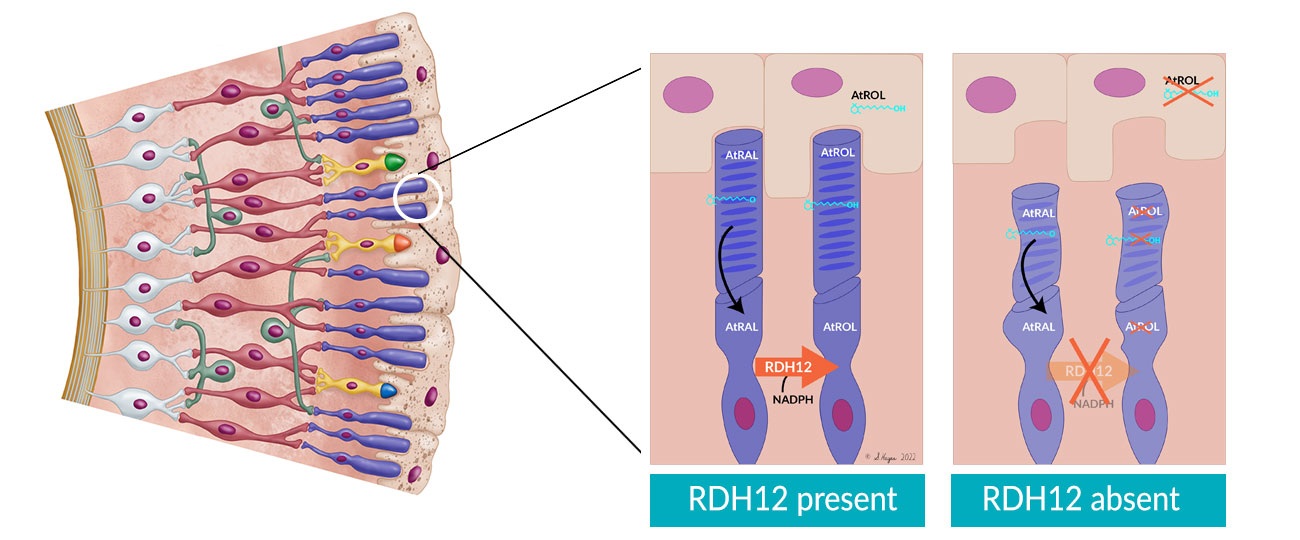
RDH12 is a severe, early-onset IRD. The RDH12 gene encodes for a retinol dehydrogenase enzyme produced in the photoreceptor inner segment that has a key role in the visual cycle. Conversion of all-trans retinal (AtRAL) to all-trans retinol (AtROL) by RDH12 during the visual cycle protects photoreceptors from cytotoxicity. Mutations in RDH12 permit a toxic accumulation of AtROL, leading to photoreceptor degeneration.
OPGx-MERTK
OPGx-MERTK is being developed for retinal degeneration caused by mutations in the MERTK gene, affecting approximately 2,600 people in the U.S. MERTK plays a critical role in phagocytosis of photoreceptor outer segments by RPE cells. Loss of this function leads to rod-cone dystrophy and progressive vision loss.
Opus is advancing OPGx-MERTK using a modern AAV vector design, building on prior preclinical proof of concept and earlier clinical experience, with the aim of improving durability and efficacy.
Preclincal programs
OPGx-NMNAT1
OPGx-NMNAT1 is a gene augmentation therapy designed to halt disease progression in pediatric patients with retinal degeneration caused by mutations in the NMNAT1 gene, which affects approximately 1,200 people in the U.S.
NMNAT1 is essential for regenerating NAD+, a critical metabolic cofactor. Preclinical data in mouse models that recapitulate key features of the human disease demonstrated that AAV-mediated delivery of NMNAT1 stabilized retinal degeneration.
OPGx-CNGB1
OPGx-CNGB1 is an AAV gene therapy program for a late-onset form of retinitis pigmentosa caused by mutations in the CNGB1 gene, affecting approximately 2,100 people in the U.S.
Opus is collaborating with an NIH-funded academic consortium and the Foundation for the NIH’s Bespoke Gene Therapy Consortium to advance OPGx-CNGB1 into and through a Phase 1 clinical trial.